|
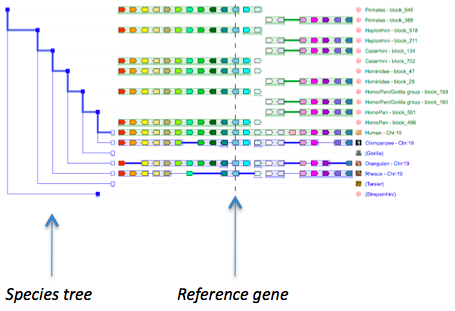
The gene initially placed in the centre of the display and aligned over a vertical black line is the gene that was used as query (reference gene).
|
|
Genes outlined in white are paralogs of the genes in the same colour but outlined in black.
|

|
PhyloView only: shaded genes correspond to genes that are not orthologous to any genes from the species used in the query (reference).
|

|
Coloured genes over a light blue background corresponds to extant or ancestral genes that are i) orthologous to genes from the species used in the query that show the same colour ii) but belong to other branches than the branch leading from the ancestral root to the extant species used as queries.
|

|
Coloured genes over a light green background corresponds to ancestral versions of the genes from the species used in the query that show the same colour
|

|
Branches shown as thicker lines in lighter blue represent the path leading from the ancestral root to the reference species used as query.
|
|
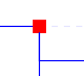
Blue square nodes represent ancestral species leading from the same "root" ancestral species to orthologs and/or paralogs of the gene used as query.
|
|
Red square nodes represent duplication events of an ancestral version of the gene used as query
|
|
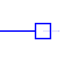
Open blue square nodes represent extant species
|



|
In PhyloView, coloured circles between genes indicate intergenic Conserved Non-coding Elements (CNEs). The colour from green to red to blue indicates the level of conservation (respectively CNE set1, set2, set3; see the
CNE section for more information)
|



|
In PhyloView, coloured bars between genes indicate intronic Conserved Non-coding Elements (CNEs). The "host" gene is always the first gene on the left of the CNE. The colour from green to red to blue indicates the level of conservation (respectively CNE set1, set2, set3; see the
CNE section for more information)
|
|
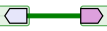
In AlignView, a thick green line between two genes in an ancestral species is equivalent to a "gap" in the alignment, i.e. the two genes are neighbours in this species but not in the reference species, where the two orthologs are separated by one or more genes.
|

|
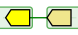
In AlignView, a thick blue line between two genes is equivalent to a "gap" in the alignment of this extant species, i.e. the two genes are neighbours in this species but not in the reference species, where their orthologs are separated by one or more genes. Clicking on the line brings up a window indicating the size of the gap in base pairs, and a link back to other genome browsers.
|
|
In AlignView, a thin green line between two genes is equivalent to a "break" in the continuity of the alignment, i.e. the two genes are linked (on the same chromosome or scaffold) in the order shown in the corresponding extant species but at least one gene separates the two genes in that species
|
|
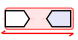
In AlignView, a thin double-headed arrow under a block of genes means that the order of the genes shown was flipped around (reversed) compared to the "canonical" orientation found in e.g. Ensembl for extant species and in this browser for ancestral species.
|